光生物催化——利用光来扩大酶的反应性,最近成为一种开发自然界新化学物质的有力策略。这些系统在不对称自由基反应中显示出了潜力,而这些反应长期以来一直无法用小分子催化剂催化。
到目前为止,非自然光催化反应仅限于整体还原和氧化还原中性过程。
在此,来自美国匹兹堡大学的刘鹏&美国加州大学圣巴巴拉分校的杨扬等研究者报道了光生物催化有机硼试剂与氨基酸之间的不对称sp3-sp3氧化交叉偶联。相关论文以题为“Stereoselective amino acid synthesis by photobiocatalytic oxidative coupling”于2024年05月01日发表在Nature上。

据悉,这早已不是二人的第一次强强联手,仅在2023年两人就曾联手发了一篇Science以及一篇Nature Catalysis。不完全统计,此前两人更联手发过,诸如JACS(2022年2篇)、Science(2021年1篇)等顶刊。

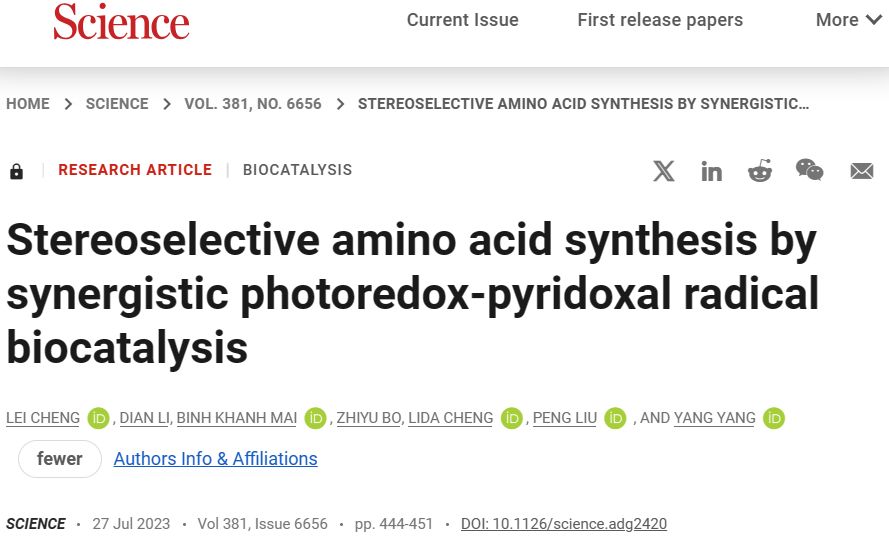
在过去的几年里,利用可见光揭示了以前虚幻的酶活性,对非自然光生物催化的研究在一系列立体选择性C-C键形成自由基过程中达到了高峰,这对手性小分子催化剂来说仍然是一个挑战。
值得注意的是,通过利用烟酰胺、黄素和吡哆醛-5 ' -磷酸依赖酶的催化乱交性,促进了整体的还原和氧化还原中性自由基转化,允许分子间自由基加成烯烃和sp3-sp3交叉亲电偶联发生,并具有良好的对映体控制。
然而,尽管亲核试剂的结构多样性和广泛可用性,两种不同的亲核试剂的氧化C(sp3)-C (sp3)交叉偶联在新的自然光生物催化中仍然未知(图1a)。如果成功实现,不对称光生物催化sp3-sp3氧化偶联将为从丰富的构建块中获得过多的增值手性产品提供方便。
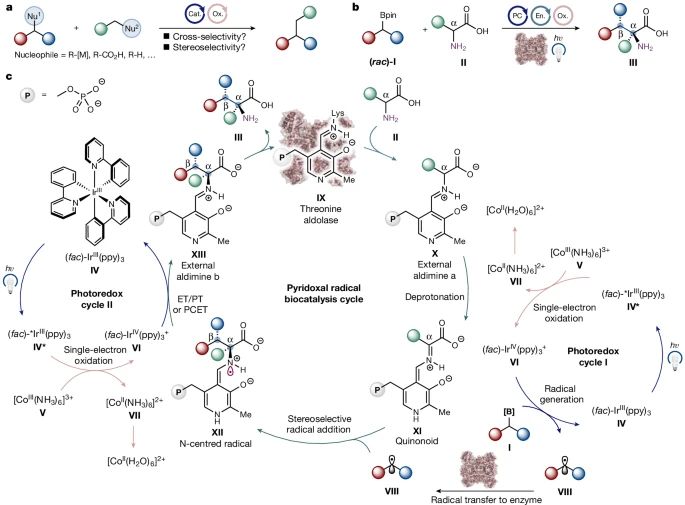
图1 光生物催化不对称sp3-sp3氧化交叉偶联的协同三催化循环。
在研究者小组设计立体选择性自由基生物催化新策略的之前努力中,研究者最近质疑是否可以推进新的生物催化激活模式来促进sp3-sp3氧化交叉偶联,这在有机合成化学或酶学中都是未知的。
鉴于天然磷酸吡哆醛(pyridoxal phosphate, PLP)依赖酶具有巨大的结构和功能多样性,研究者对解锁广泛使用的PLP生物催化剂的新的氧化自由基偶联活性特别感兴趣,这些生物催化剂用于立体选择性合成非规范氨基酸(ncAAs)。
ncAAs广泛应用于临床重要的肽治疗、生物活性天然产物和功能性非天然蛋白质中,是一类有价值的化合物,其立体选择性合成长期以来一直吸引着合成化学家和合成生物学家。在自然界中,PLP酶通过羰基催化作用,在氨基酸底物的α、β和γ位置通过双电子机制促进多种ncAAs的构建和破坏。
直到最近,自由基PLP酶仅限于天然的[4Fe-4S]/SAM和B12依赖的PLP氨基转化酶和O2依赖的PLP氧化酶,底物范围很窄。在2023年,研究者的团队描述了一种新的非自然光生物催化形式,其中研究者利用可见光光氧化还原催化剂和酶的合作相互作用来开启PLP依赖性色氨酸合成酶的非自然自由基反应活性,用于丝氨酸和苏氨酸的β-去羟基交叉偶联。
研究者假设,如果另一个不同的α-功能化PLP酶家族,包括苏氨酸醛缩酶、丝氨酸羟甲基转移酶、苏氨酸转醛缩酶(TTAs)及其相关的生物合成酶,可以被重新利用并进化为sp3-sp3氧化偶联,这将使研究者能够推进一种前所未有的吡啶醛自由基生物催化模式,用于立体选择性和无保护基团的ncAAs合成。包括那些带有连续立体中心和α-四取代立体中心的,这些仍然是催化不对称合成所不能达到的(图1b)。
在此,研究者提出的通过光氧化还原-吡哆醛生物催化(I + II→III)进行sp3-sp3氧化偶联的三重催化循环,详细描述如图1c所示。在可见光照射下,(fac)-Ir(ppy)3 (ppy = 2-苯基吡啶,IV)会吸收一个光子,提供一个长寿命的激发态还原剂(fac)-*Ir(ppy)3 (IV*,E1/2 (*IrIII/IrIV) = - 1.73 V,相对于CH3CN中的饱和甘汞电极(SCE))。
(fac)-*Ir(ppy)3将被[Co(NH3)6]3+ (V, E1/2 (CoIII/CoII)与SCE在H2O)中迅速氧化,得到(fac)-Ir(ppy)3+ (VI)作为强氧化剂(E1/2 (IrIV/IrIII=0.77 V),相对于CH3CN中的SCE。由于还原后的[Co(NH3)6]2+ (VII)的配体稳定性增加,与H2O的快速配体交换将形成[Co(H2O)6]2+ (E1/2 (CoIII/CoII) = 1.68 V,相对于H2O中的SCE)中不能被(fac)-Ir(ppy)3+ (VI)再氧化。
有机硼底物(I)与VI的单电子氧化,将提供瞬态碳中心自由基VIII并再生(fac)-Ir(ppy)3光催化剂(IV)。在生物催化循环中,依赖PLP的α-功能化酶IX,如苏氨酸醛缩酶,首先与丰富的氨基酸底物II进行传递,形成外部醛胺X。
PLP结合会导致α-质子的显著pKa降低,从而导致关键的类醌中间体(XI)的快速去质子化。在这个关键阶段,光催化形成的自由基物种VIII会进入酶的活性位点,并立体选择性地添加到酶形成的类醌XI的Cα碳上,从而形成氮中心自由基XII,这是原生PLP酶学中难以找到的物种。
(fac)-Ir(ppy)3+单电子氧化XII生成新的外醛胺XIII,该外醛胺XIII随后释放出sp3-sp3偶联的ncAA产物III并再生PLP生物催化剂IX,从而完成所有催化循环。
如果成功进行,这种氧化C(sp3) -C (sp3)交叉偶联将使两种不同的亲核试剂结合,包括容易获得的有机硼试剂和丰富的氨基酸底物,通过C(sp3) -H功能化逻辑。
此外,如果酶控制的自由基加成和质子转移步骤具有良好的立体选择性,则该α-吡哆醛自由基生物催化将允许制备具有两个相邻立体中心的不同ncAAs,具有良好的非立体和对映控制。
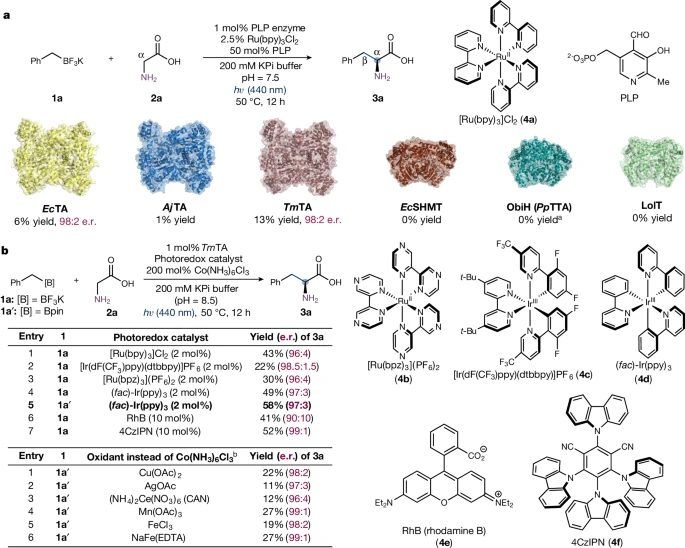
图2 光生物催化不对称sp3-sp3氧化交叉偶联的发现与优化。
在本研究开始时,研究者着手确定合适的PLP生物催化剂,以促进所提出的三氟硼酸苄酯(1a)和甘氨酸(2a,图2)的光生物催化氧化交叉偶联。原则上,绝大多数能够形成必要的类醌中间体(XI)的PLP依赖酶都可以被重新利用,以允许氨基酸的自由基α-功能化。
最终,在研究者评价的单电子氧化剂中,Mn(III), Fe(III)和Co(III)盐有效地促进了sp3-sp3偶联。最终发现,Co(NH3)6Cl3是最佳的氧化剂。在此条件下,苯三氟硼酸酯1a和苯硼酸松醇酯1a′均能有效转化,且当1a′与2mol % (fac)-Ir(ppy)3 (4d)偶联时,产率提高。
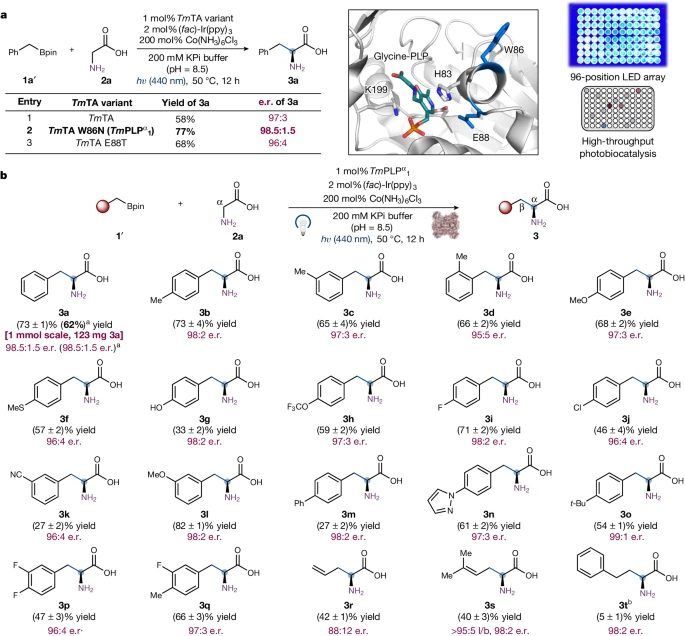
图3 光生物催化不对称sp3-sp3氧化偶联合成ncAA。
值得注意的是,无需进一步的酶工程,烯丙基硼酸盐(3r和3s)可以转化为具有良好对映体比例的相应的烯基ncAAs。其中,丙烯基硼酸酯(3s)的线性(l)与支链(b)的转化比优异(大于95:5 l:b)。当罗丹明B (4e)作为光催化剂时,非活化的烷基硼酸盐(3t)也被酶所接受,使ncAA产物具有良好的对映选择性,尽管产率很低。
总之,这些结果表明α-吡哆醛自由基生物催化,在进一步定向进化中具有适应广泛有机硼试剂的良好潜力。重要的是,这种光催化氨基酸合成被发现是可扩展的。研究者观察到在10.0 mM的1a '浓度下成功制备了1 mmol规模的3a,而没有降低生物转化的对映体选择性。
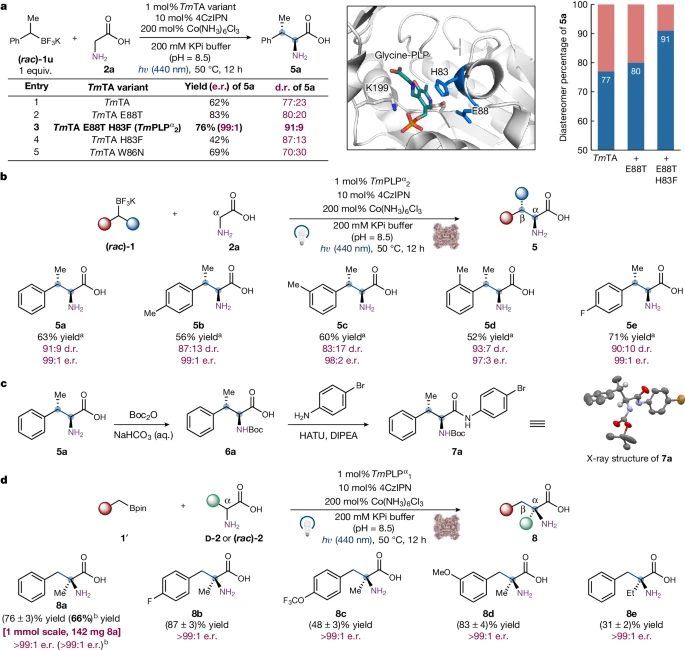
图4 β-甲基ncAAs的对映和非对映选择性合成和α-四取代ncAAs的对映选择性合成。
研究者测试了他们的PLP自由基酶,使用外消旋仲烷基硼酸盐以对映趋同的方式形成连续的立体中心(图4)。β-甲基ncAAs包含了一个新兴的但在很大程度上未知的肽治疗化学空间,因为含有这些结构元件的肽具有增强的蛋白水解稳定性和结合亲和力。在不操纵保护基团的情况下高效催化不对称合成邻α和β-立体中心的ncAAs是药物化学家面临的一项重要任务。
使用外消旋的1-苯乙基三氟硼酸盐1u作为自由基前体,研究者发现野生型TmTA能够通过氧化C-C偶联催化合成β-甲基ncAAs,以62%的产率,77:23的非对映比(d.r)和99:1的e.r递送(2S,3S)-5a(图4a)。与伯烷基硼试剂的氧化偶联不同,对于仲烷基亲核试剂,烷基三氟硼酸盐和4CzIPN的使用,相对于烷基硼酸蒎醇酯和(fac)-Ir(ppy)3的产率更高。
为了提高这种对映收敛性氧化sp3-sp3交叉偶联的催化效率和非对映选择性,研究者使用反复的位点饱和诱变(SSM)和研究者内部构建的高通量光化学设备进行了TmTA的定向进化(图4a)。
通过两轮定点饱和突变(SSM)和筛选,TmTA E88T H83F (TmPLPα2)以关键α-螺旋上的88和83位残基为靶点,进化出(2S,3S)-5a,产量为76%,d.r为91:9。在引入的两个有益突变中,E88T增强了酶活性,而H83F改善了非对异调控。利用具有对(5b)、间(5c)、邻甲基(5d)和氟(5e)的TmPLPα2,1-苯乙基三氟硼酸酯,可以成功转化为β-甲基苯丙氨酸类似物,收率高,非映对和对映控制效果好(图4b)。
进一步的研究表明,没有发生(rac)-1u的动力学分解,证实了这种生物转化的对映收敛性质。通过对其酰胺衍生物7a的单晶X射线衍射分析,确定了5a的绝对立体化学和相对立体化学(图4c)。总之,这种光生物催化氧化交叉偶联在一次操作中提供了聚合、立体选择性和无保护基团的有价值的β-甲基ncAAs合成。
综上所述,协同光氧化还原-吡哆醛生物催化,为sp3-sp3氧化偶联提供了一个平台,允许化学或生物学未知的立体选择性、分子间自由基转化。
【参考文献】
Wang, TC., Mai, B.K., Zhang, Z. et al. Stereoselective amino acid synthesis by photobiocatalytic oxidative coupling. Nature 629, 98–104 (2024).
