本文华算科技介绍了常见的吸附位点、桥位、阶边等),并从计算化学角度说明如何通过DFT几何优化、吸附能文中还给出建模、色散校正、覆盖度与热力学修正等实践建议在固体表面或纳米材料上,分子或原子“停靠”的位置并不只有一个,而是有多种典型位点,常见的有(on-top)、(bridge)和(hollow)。
就是直接坐落在表面单个原子正上方,几何最直观;位于两个表面原子之间的连线上;通常位于三个或四个基元原子环中心,按晶格可分为fcc/hcp等不同的几何类型。下图展示了Cu(100)表面终止层的三种化学吸附位点示意图。
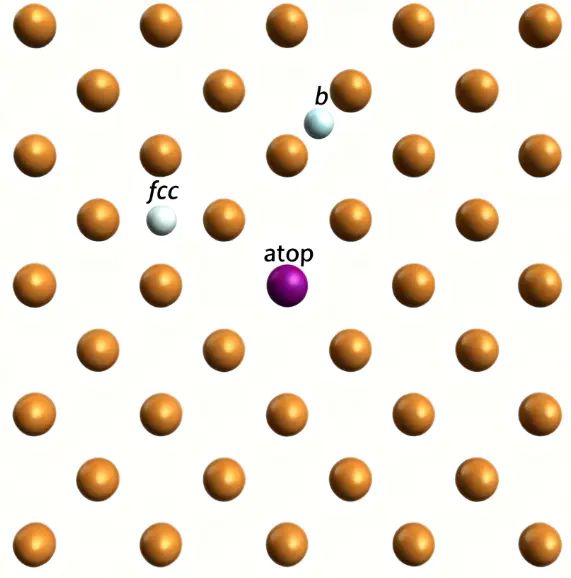
10.1039/D3CP04021F
台阶阶边台阶角()点缺陷此外,对于多孔材料、纳米簇或分子表面,还有内部孔道位点、配位位点(如金属位点)、配体位点(有机官能团)与双位点/二齿位(bidentate)等,吸附物可能跨越两个相邻原子形成复杂结合模式总之,取决于材料的结构(氧化态、表面修饰),理解这些基本类型是判断吸附行为的起点。
如何识别与评估位点优先级
开始,然后通过几何优化得到最稳构型。
Eads = E(表面+吸附物) − E(表面) − E(吸附物),负值越大表示吸附越DFT:投影态密度(PDOS)显示键形成的轨道贡献,电荷分析(如Bader)揭示电子转移,电荷密度差(Δρ)图直观显示成键/反键区域。
(top、bridge、fcc和hcp)的示意图,同时进一步绘制了平均CO吸附能随覆盖度(0.25 ML至1.0 ML)的变化曲线,显示吸附能从约-1.4 eV降至-0.5 eV,体现了横向排斥作用的影响。
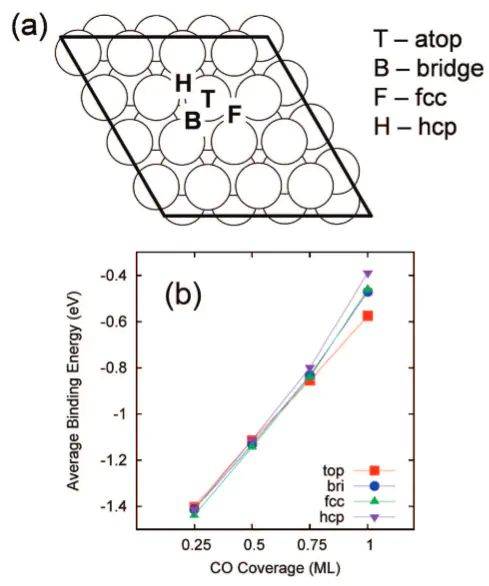
,可通过(化学吸附会有明显的新振动模)来区分。
位点能垒与迁移行为NEB实际计算还要注意覆盖度效应(电极界面)对位点能量的显著影响,因此在不同条件下反复比较位点的自由能比单纯的势能更能反映实验可达性。
CI-NEB2+Na。
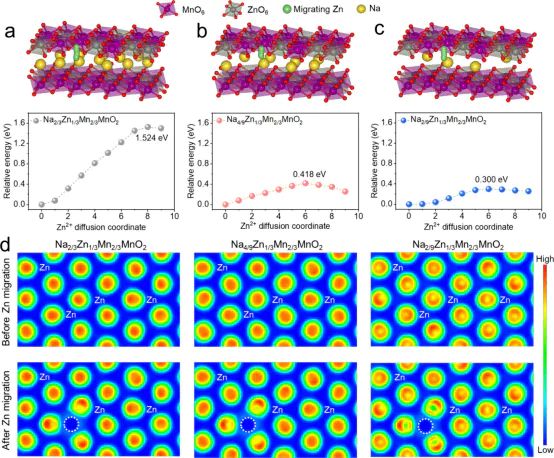
10.1021/acsenergylett.4c03136
实践建议
初学者的实操建议1)不要只优化一个位点就下结论,系统地测试顶、桥、空心、台阶与缺陷等多种位点2)选用合适的,并做k点、超胞尺寸和真空厚度的收敛测试;若体系涉及弱吸附,务必校验色散贡献和BSSE影响。
用于零点能和热力学修正,可得到吸附自由能以便与TPD或热力学实验比对。对于电化学体系,考虑恒电势或电荷模型以对齐实验电位。
XPS:谱学峰位、脱附温度和表面形貌是判定位点类型与活性的关键证据。
,为催化设计、传感器优化或吸附材料工程提供坚实依据。
总结
/局部几何和电子环境不同计算化学提供了一套从定位位点到评估热力学与动力学可行性的工具:
DFT用于吸附能和电子结构分析,振动计算用于自由能修正与谱学对比将系统化的多位点筛选与实验数据结合